How does each gene’s noncoding regulatory DNA determine its expression?
When a cell switches from performing one set of functions to another, either in development or in response to a changing external environment, many thousands of genes are regulated, altering their expression, their chromatin structure, and even how they are packed and positioned within the nucleus. Understanding the genetic and molecular basis of development therefore demands a quantitative description of how each gene is regulated. Sequencing is routinely used to probe various layers of cells’ molecular state, but a quantitative epigenetic model of how chromatin accessibility, transcription factor binding, histone modifications, DNA methylation, and other “inputs” determine a gene’s transcriptional output remains elusive.
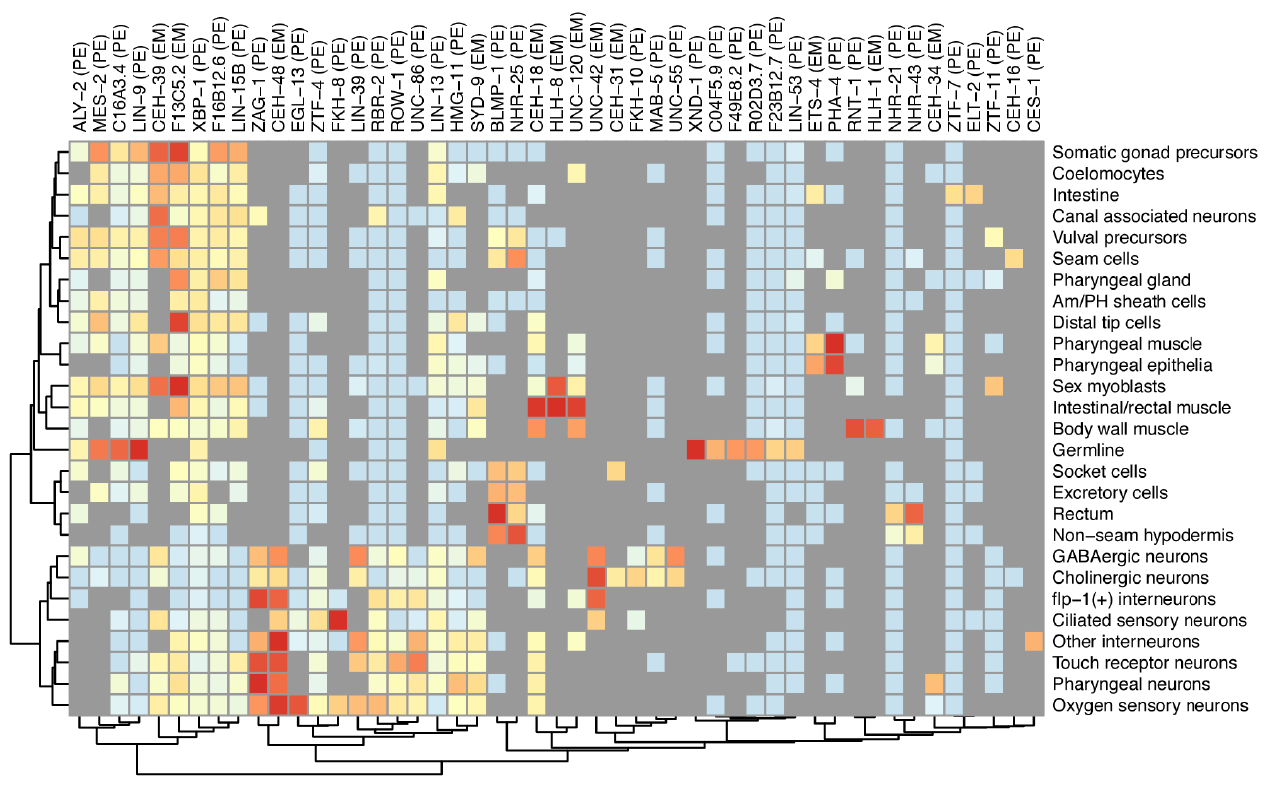
We aim to use single-cell genomics experiments to train statistical models that can explain how every gene is regulated in every cell in an entire animal. Our models integrate DNA sequence and epigenetic measurements such as chromatin accessibility to predict a gene’s expression. We’ve recently devised techniques that link noncoding regulatory DNA to target genes and describe how their chromatin accessibility drives gene regulation. We were also able to construct an organism-scale model that quantifies each of 235 different C. elegans transcription factors’ contribution to establishing the specific expression profile of 27 different cell types. As we develop new or improved single-cell assays based on combinatorial cellular indexing, we will expand the scope and power these models and those describing other systems used in the lab.