Abstract
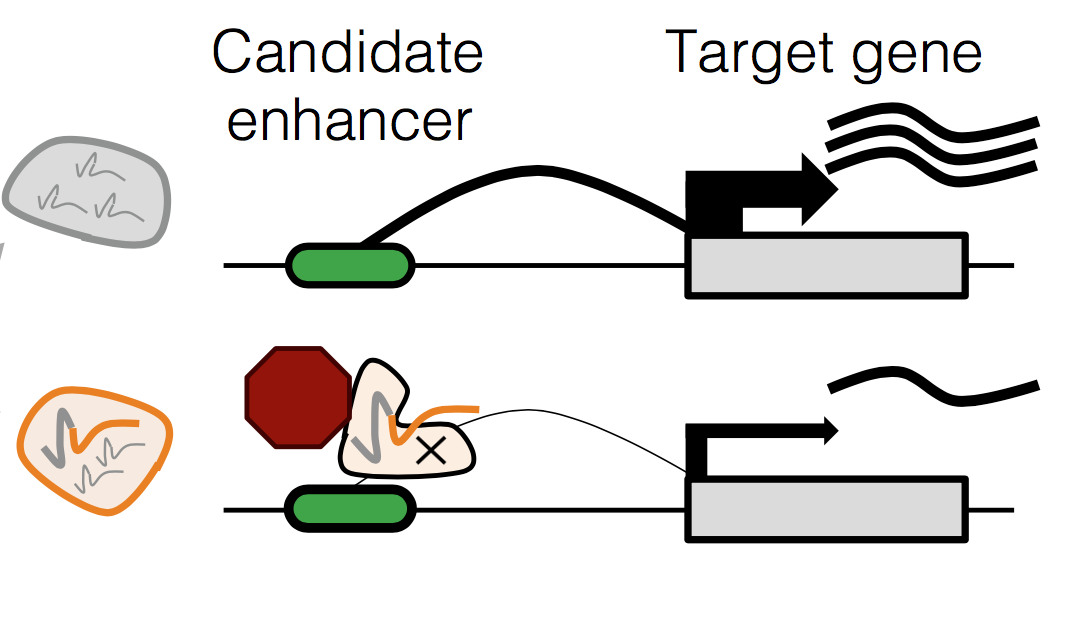
Over one million candidate regulatory elements have been identified across the human genome, but nearly all are unvalidated and their target genes uncertain. Approaches based on human genetics are limited in scope to common variants and in resolution by linkage disequilibrium. We present a multiplex, expression quantitative trait locus (eQTL)-inspired framework for mapping enhancer-gene pairs by introducing random combinations of CRISPR/Cas9-mediated perturbations to each of many cells, followed by single-cell RNA sequencing (RNA-seq). Across two experiments, we used dCas9-KRAB to perturb 5,920 candidate enhancers with no strong a priori hypothesis as to their target gene(s), measuring effects by profiling 254,974 single-cell transcriptomes. We identified 664 (470 high-confidence) cis enhancer-gene pairs, which were enriched for specific transcription factors, non- housekeeping status, and genomic and 3D conformational proximity to their target genes. This framework will facilitate the large-scale mapping of enhancer-gene regulatory interactions, a critical yet largely uncharted component of the cis-regulatory landscape of the human genome.