Abstract
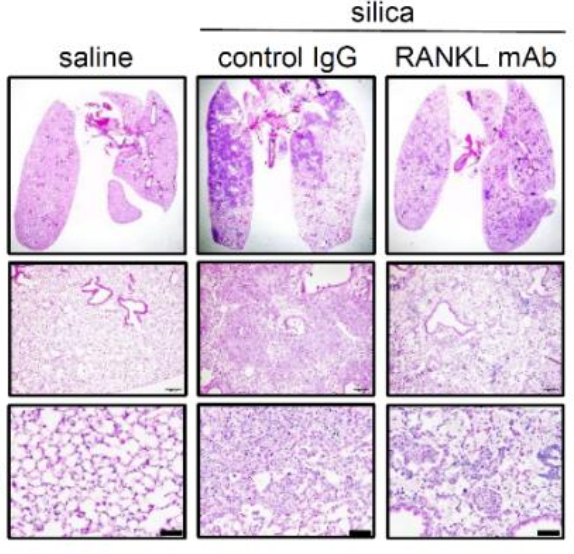
The pathophysiology of silicosis is poorly understood, limiting development of therapies for those who have been exposed to the respirable particle. We explored the mechanisms of silica-induced pulmonary fibrosis in a mouse model using multiple modalities including wholelung single-nucleus RNA sequencing. These analyses revealed that in addition to pulmonary inflammation and fibrosis, intratracheal silica challenge induced osteoclast-like differentiation of alveolar macrophages and recruited monocytes, driven by induction of the osteoclastogenic cytokine, receptor activator of nuclear factor-κB ligand (RANKL) in pulmonary lymphocytes and alveolar type II cells. Furthermore, anti-RANKL monoclonal antibody treatment suppressed silica-induced osteoclast-like differentiation in the lung and attenuated silica-induced pulmonary fibrosis. We conclude that silica induces osteoclast-like differentiation of distinct recruited and tissue resident monocyte populations, leading to progressive lung injury, likely due to sustained elaboration of bone resorbing proteases and hydrochloric acid. Interrupting osteoclast-like differentiation may therefore constitute a promising avenue for moderating lung damage in silicosis.
One Sentence Summary: Silica induces the alveolar epithelium to reprogram recruited and resident pulmonary myeloid cells to become osteoclasts that contribute to pulmonary fibrosis.
# equal contribution ** corresponding authors