Abstract
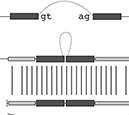
Motivation: A new protocol for sequencing the messenger RNA in a cell, known as RNA-Seq, generates millions of short sequence fragments in a single run. These fragments, or ‘reads’, can be used to measure levels of gene expression and to identify novel splice variants of genes. However, current software for aligning RNA-Seq data to a genome relies on known splice junctions and cannot identify novel ones. TopHat is an efficient read-mapping algorithm designed to align reads from an RNA-Seq experiment to a reference genome without relying on known splice sites.
Results: We mapped the RNA-Seq reads from a recent mammalian RNA-Seq experiment and recovered more than 72% of the splice junctions reported by the annotation-based software from that study, along with nearly 20 000 previously unreported junctions. The TopHat pipeline is much faster than previous systems, mapping nearly 2.2 million reads per CPU hour, which is sufficient to process an entire RNA-Seq experiment in less than a day on a standard desktop computer. We describe several challenges unique to ab initio splice site discovery from RNA-Seq reads that will require further algorithm development.
Availability: TopHat is free, open-source software available from http://tophat.cbcb.umd.edu