The Cufflinks RNA-Seq workflow
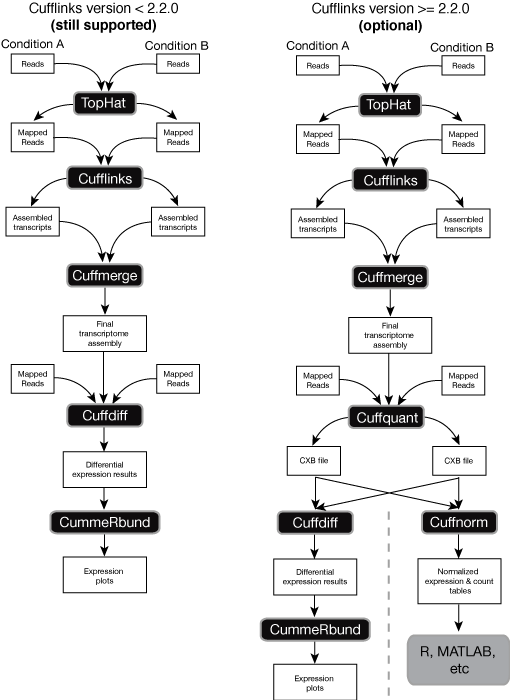
The Cufflinks suite of tools can be used to perform a number of different types of analyses for RNA-Seq experiments. The Cufflinks suite includes a number of different programs that work together to perform these analyses. The complete workflow, performing all the types of analyses Cufflinks can execute, is summarized in the graph below. The left side illustrates the “classic” RNA-Seq workflow, which includes read mapping with TopHat, assembly with Cufflinks, and visualization and exploration of results with CummeRbund. A newer, more advanced worfklow was introduce with Cufflinks version 2.2.0, and is shown on the right. Both are still supported. You can read about the classic workflow in detail in our protocol paper.
Cufflinks
Cufflinks is both the name of a suite of tools and a program within that suite. Cufflinks the program assembles transcriptomes from RNA-Seq data and quantifies their expression.
Cuffcompare
After assembling a transcriptome from one or more samples, you’ll probably want to compare your assembly to known transcripts. Even if there is no “reference” transcriptome for the organism you’re studying, you may want to compare the transcriptomes assembled from different RNA-Seq libraries. Cuffcompare helps you perform these comparisons and assess the quality of your assembly.
Cuffmerge
When you have multiple RNA-Seq libraries and you’ve assembled transcriptomes from each of them, we recommend that you merge these assemblies into a master transcriptome. This step is required for a differential expression analysis of the new transcripts you’ve assembled. Cuffmerge performs this merge step.
Cuffquant
Quantifying gene and transcript expression in RNA-Seq samples can be computationally expensive. Cuffquant allows you to compute the gene and transcript expression profiles and save these profiles to files that you can analyze later with Cuffdiff or Cuffnorm. This can help you distribute your computational load over a cluster and is recommended for analyses involving more than a handful of libraries.
Cuffdiff
Comparing expression levels of genes and transcripts in RNA-Seq experiments is a hard problem. Cuffdiff is a highly accurate tool for performing these comparisons, and can tell you not only which genes are up- or down-regulated between two or more conditions, but also which genes are differentially spliced or are undergoing other types of isoform-level regulation.
Cuffnorm
Sometimes, all you want to do is normalize the expression levels from a set of RNA-Seq libraries so that they’re all on the same scale, facilitating downstream analyses such as clustering. Expression levels reported by Cufflinks in FPKM units are usually comparable between samples, but in certain situations, applying an extra level of normalization can remove sources of bias in the data. Cuffnorm normalizes a set of samples to be on as similar scales as possible, which can improve the results you obtain with other downstream tools.
File Formats
The Cufflinks suite of tools report output in a number of different file formats. While some of these are standard, others are unique, and they are described here.