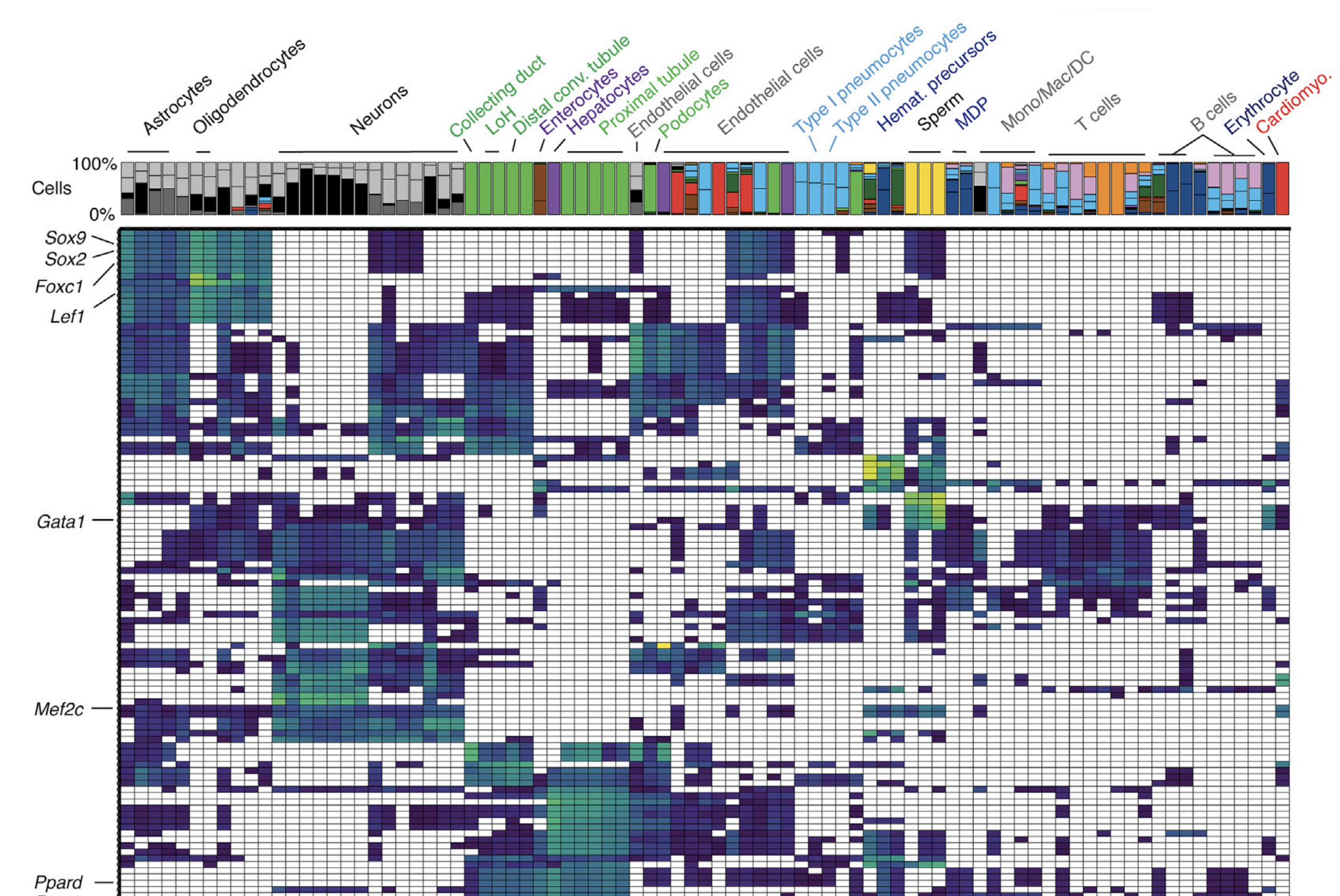
How does the genome encode cells’ ability to specialize into so many different types? Mammals contain many different types of cells, each of which has a distinct program of gene expression. The particular genes expressed in each type of cell are believed to be determined in part by which noncoding regulatory sequences in the genome can be accessed by transcription factors and other regulatory proteins. However, the chromatin accessibility profiles for most cell types remain undefined, and the gene targets for the overwhelming majoriry of regulatory DNA sequences are unknown.
In collaboration with the Jay Shendure’s lab, we recently measured chromatin accessibility in more than 100,000 individual cells in the adult mouse, deriving accessibility profiles for 85 different cell types. We developed a new algorithm, Cicero, to link accessibile elements to their target genes. We also tracked the dynamics of chromatin accessibility during differentiation of blood cells using Monocle. Finally, we were able to use deep learning to build models of how DNA sequence controls the patterns of accessibility unique to each cell type. As we integrate these data with single-cell RNA-seq analyses of the mouse from our group and others, we hope to derive a better understanding of how the genome encodes each cell’s types particular program of gene regulation.
You can dive into our data and build on or modify our analysis code, both of which are freely available.