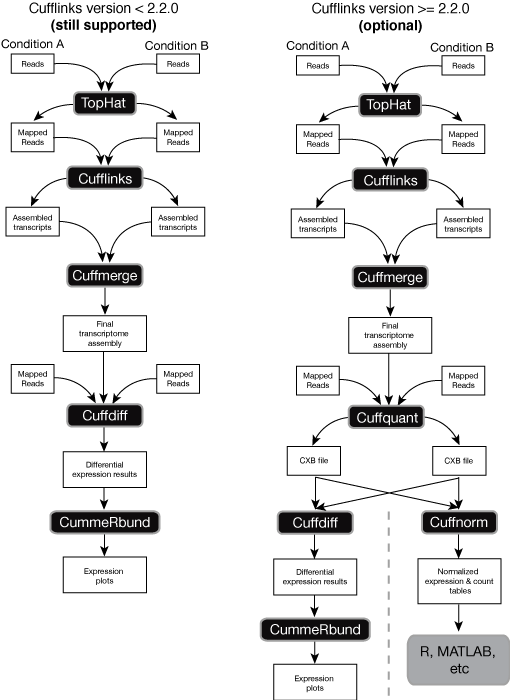
This release introduces some new features designed to simplify and speed up Cufflinks workflows. Release version 2.2.0 includes two new programs, Cuffquant and Cuffnorm that make it easier to quantify gene expression in experiments with many samples. These are particularly helpful for single cell RNA-Seq experiments, where the reads for each cell are provided as a separate FASTQ file or pair of files.
Release 2.2.0 also introduces sample sheets and contrast files(manual.html). These facilities make it easier to work with large analyses involving many samples.
Cuffquant quantifies gene and transcript expression levels for a single BAM file. These levels are stored in a new binary file type, the CXB file. You can then provide CXB files for your samples directly to Cuffdiff instead of BAMs. Mixing BAM and CXB files is not yet supported. Because expression levels for each sample are quantified by Cuffquant, Cuffdiff doesn’t have to perform this step, which speeds up Cuffdiff runs substantially and lowers their memory footprints. We recommend that most users switch to this new Cuffdiff workflow for all experiments that involve more than a few samples. However, note that running Cuffquant prior to running Cuffdiff is optional - you can still directly supply BAM files to Cuffdiff.
Cuffquant files can also be passed to Cuffnorm, which simply computes a normalized table of expression values for genes and transcripts. Unlike Cuffdiff, Cuffnorm performs no differential expression testing. Cuffnorm also does not calculate confidence intervals on expression values. Cuffnorm is meant to be used when all you want is a set of comparable expression values for genes, transcripts, CDS groups, and TSS groups, such as when you simply want to make heatmaps or dendrograms or plot values for individual genes. Cuffnorm can output values in one of two formats: Cuffdiff tracking tables (which can be used with CummeRbund), or in simple, tab-delimited tables. The latter is the recommended output format for use with Monocle in single cell RNA-Seq experiments.
Here’s an overview of the new (optional) workflow, compared to the old workflow described in our Nature Protocols paper: