Cicero is an ongoing research project as well as a toolkit.
If you use Cicero, please cite these papers in your work!
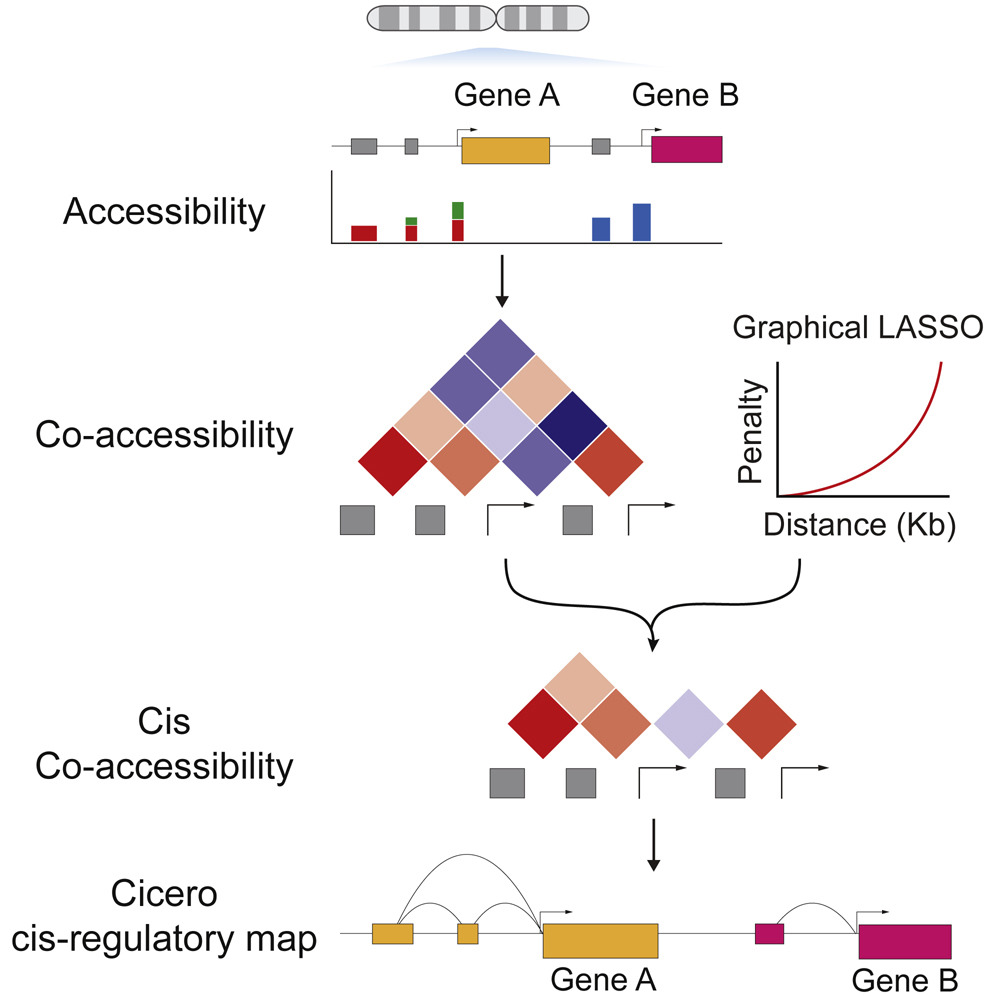
Cicero Predicts cis-Regulatory DNA Interactions from Single-Cell Chromatin Accessibility Data
Hannah A. Pliner, Jonathan S. Packer, José L. McFaline-Figueroa, Darren A. Cusanovich, Riza M. Daza, Sanjay Srivatsan, Xiaojie Qiu, Dana Jackson, Anna Minkina, Andrew C. Adey, Frank J. Steemers, Jay Shendure*, Cole Trapnell*.
Molecular Cell 71, 1–14 2018
Linking regulatory DNA elements to their target genes, which may be located hundreds of kilobases away, remains challenging. Here, we introduce Cicero, an algorithm that identifies co-accessible pairs of DNA elements using single-cell chromatin accessibility data and so connects regulatory elements to their putative target genes. We apply Cicero to investigate how dynamically accessible elements orchestrate gene regulation in differentiating myoblasts. Groups of Cicero-linked regulatory elements meet criteria of “chromatin hubs”—they are enriched for physical proximity, interact with a common set of transcription factors, and undergo coordinated changes in histone marks that are predictive of changes in gene expression. Pseudotemporal analysis revealed that most DNA elements remain in chromatin hubs throughout differentiation. A subset of elements bound by MYOD1 in myoblasts exhibit early opening in a PBX1- and MEIS1-dependent manner. Our strategy can be applied to dissect the architecture, sequence determinants, and mechanisms of cis-regulation on a genome-wide scale.
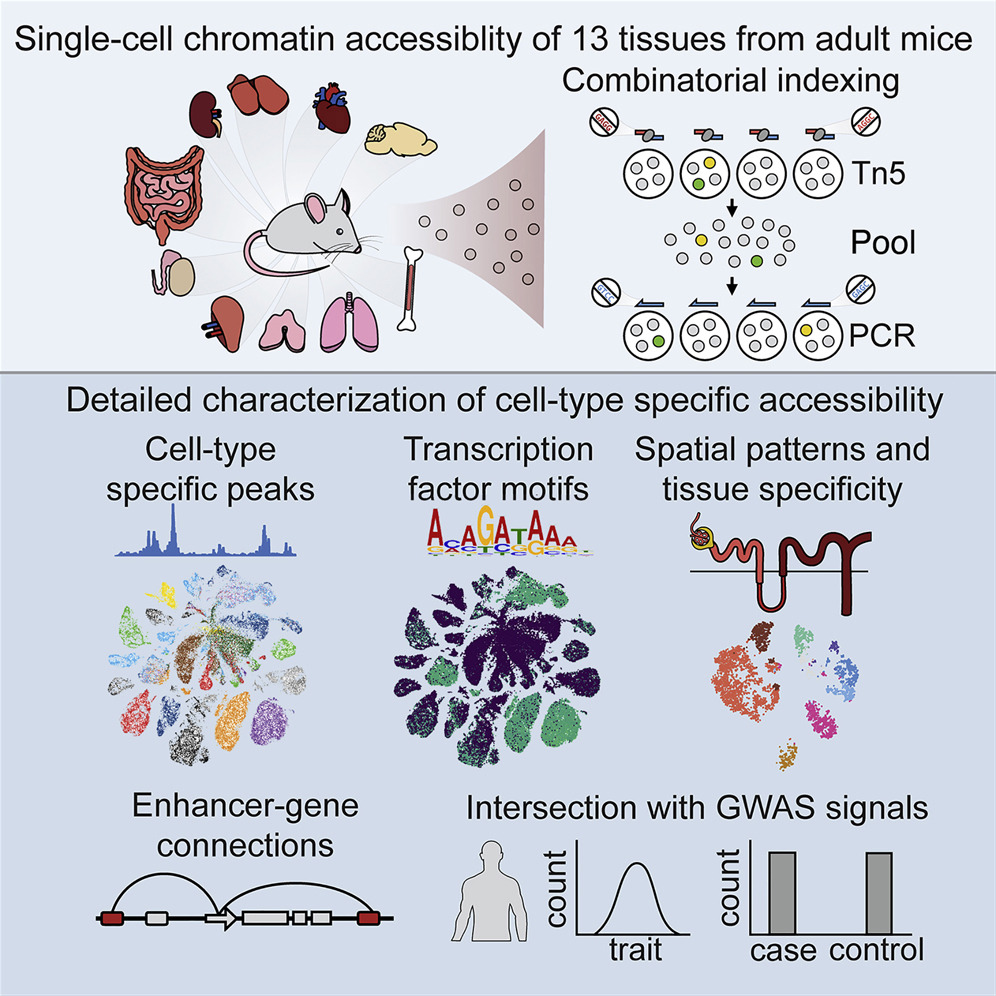
A Single-Cell Atlas of In Vivo Mammalian Chromatin Accessibility
Darren A. Cusanovich, Andrew J. Hill, Delasa Aghamirzaie, Riza M. Daza, Hannah A. Pliner, Joel B. Berletch, Galina N. Filippova, Xingfan Huang, Lena Christiansen, William S. DeWitt, Choli Lee, Samuel G. Regalado, David F. Read, Frank J. Steemers, Christine M. Disteche, Cole Trapnell, Jay Shendure.
Cell 174, 1–16 2018
We applied a combinatorial indexing assay, sci-ATAC-seq, to profile genome-wide chromatin accessibility in ∼100,000 single cells from 13 adult mouse tissues. We identify 85 distinct patterns of chromatin accessibility, most of which can be assigned to cell types, and ∼400,000 differentially accessible elements. We use these data to link regulatory elements to their target genes, to define the transcription factor grammar specifying each cell type, and to discover in vivo correlates of heterogeneity in accessibility within cell types. We develop a technique for mapping single cell gene expression data to single-cell chromatin accessibility data, facilitating the comparison of atlases. By intersecting mouse chromatin accessibility with human genome-wide association summary statistics, we identify cell-type-specific enrichments of the heritability signal for hundreds of complex traits. These data define the in vivo landscape of the regulatory genome for common mammalian cell types at single-cell resolution.