Constructing single-cell trajectories
During development, in response to stimuli, and throughout life, cells transition from one functional "state" to another. Cells in different states express different sets of genes, producing a dynamic repetoire of proteins and metabolites that carry out their work. As cells move between states, they undergo a process of transcriptional re-configuration, with some genes being silenced and others newly activated. These transient states are often hard to characterize because purifying cells in between more stable endpoint states can be difficult or impossible. Single-cell RNA-Seq can enable you to see these states without the need for purification. However, to do so, we must determine where each cell is in the range of possible states.
Monocle introduced the strategy of using RNA-Seq for single-cell trajectory analysis. Rather than purifying cells into discrete states experimentally, Monocle uses an algorithm to learn the sequence of gene expression changes each cell must go through as part of a dynamic biological process. Once it has learned the overall "trajectory" of gene expression changes, Monocle can place each cell at its proper position in the trajectory. You can then use Monocle's differential analysis toolkit to find genes regulated over the course of the trajectory, as described in the section Finding genes that change as a function of pseudotime . If there are multiple outcomes for the process, Monocle will reconstruct a "branched" trajectory. These branches correspond to cellular "decisions", and Monocle provides powerful tools for identifying the genes affected by them and involved in making them. You can see how to analyze branches in the section Analyzing branches in single-cell trajectories .
The workflow for reconstructing trajectories is very similar to the workflow for clustering, but it has a few additional
steps. To illustrate the workflow, we will use another
expression_matrix <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_expression.rds"))
cell_metadata <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_colData.rds"))
gene_annotation <- readRDS(url("https://depts.washington.edu:/trapnell-lab/software/monocle3/celegans/data/packer_embryo_rowData.rds"))
cds <- new_cell_data_set(expression_matrix,
cell_metadata = cell_metadata,
gene_metadata = gene_annotation)Pre-process the data
Pre-processing works exactly as in clustering analysis. This time, we will use a different strategy for batch correction, which includes what
Note: Your data will not have the loading batch information demonstrated here, you will correct batch using your own batch information.
cds <- preprocess_cds(cds, num_dim = 50)
cds <- align_cds(cds, alignment_group = "batch", residual_model_formula_str = "~ bg.300.loading + bg.400.loading + bg.500.1.loading + bg.500.2.loading + bg.r17.loading + bg.b01.loading + bg.b02.loading")Note that in addition to using the alignment_group argument to align_cds(), which aligns groups of cells (i.e. batches), we are also using residual_model_formula_str. This argument is for subtracting continuous effects. You can use this to control for things like the fraction of mitochondrial reads in each cell, which is sometimes used as a QC metric for each cell. In this experiment (as in many scRNA-seq experiments), some cells spontanously lyse, releasing their mRNAs into the cell suspension immediately prior to loading into the single-cell library prep. This "supernatant RNA" contaminates each cells' transcriptome profile to a certain extent. Fortunately, it is fairly straightforward to estimate the level of background contamination in each batch of cells and subtract it, which is what Packer et al did in the original study. Each of the columns bg.300.loading, bg.400.loading, corresponds to a background signal that a cell might be contaminated with. Passing these colums as terms in the residual_model_formula_str tells align_cds() to subtract these signals prior to dimensionality reduction, clustering, and trajectory inference. Note that you can call align_cds() with alignment_group, residual_model_formula, or both.
Reduce dimensionality and visualize the results
Next, we reduce the dimensionality of the data. However, unlike clustering, which works well with both UMAP and t-SNE, here we strongly urge you to use UMAP, the default method:
cds <- reduce_dimension(cds)
plot_cells(cds, label_groups_by_cluster=FALSE, color_cells_by = "cell.type")As you can see, despite the fact that we are only looking at a small slice of this dataset, Monocle reconstructs a trajectory with numerous branches. Overlaying the manual annotations on the UMAP reveals that these branches are principally occupied by one cell type.
As with clustering analysis, you can use plot_cells() to visualize how individual genes vary along the
trajectory. Let's look at some genes with interesting patterns of expression in ciliated neurons:
ciliated_genes <- c("che-1",
"hlh-17",
"nhr-6",
"dmd-6",
"ceh-36",
"ham-1")
plot_cells(cds,
genes=ciliated_genes,
label_cell_groups=FALSE,
show_trajectory_graph=FALSE)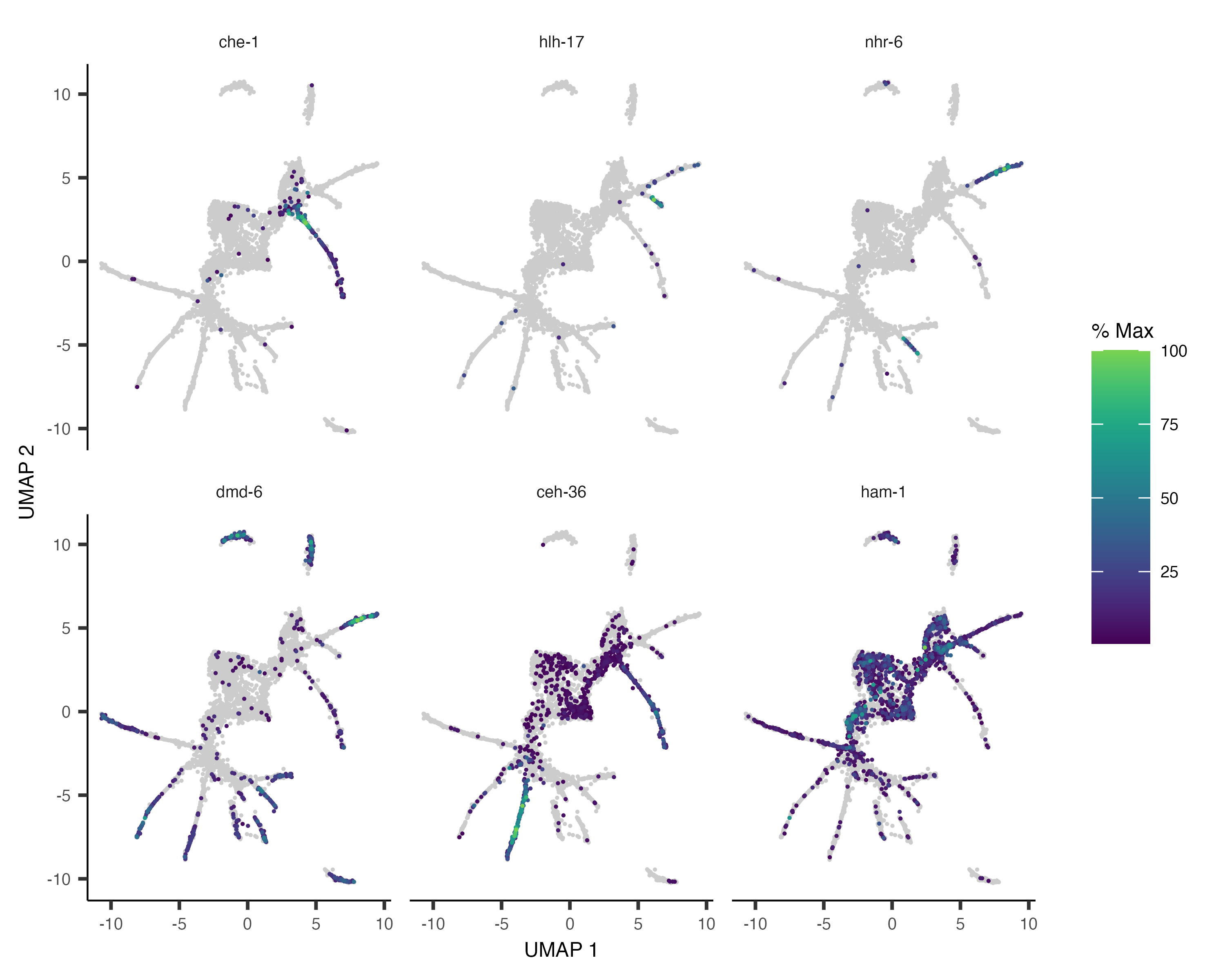
We will learn how to identify the genes that are restricted to each outcome of the trajectory later on in the section Finding genes that change as a function of pseudotime.
Cluster your cells
Although cells may continuously transition from one state to the next with no discrete boundary between them, Monocle does not assume that all cells in the dataset descend from a common transcriptional "ancestor". In many experiments, there might in fact be multiple distinct trajectories. For example, in a tissue responding to an infection, tissue resident immune cells and stromal cells will have very different initial transcriptomes, and will respond to infection quite differently, so they should be a part of the same trajectory.
Monocle is able to learn when cells should be placed in the same trajectory as opposed to separate trajectories through
its clustering procedure. Recall that we run cluster_cells(), each cell is assigned not only to a cluster
but also to a cluster_cells()as before.
cds <- cluster_cells(cds)
plot_cells(cds, color_cells_by = "partition")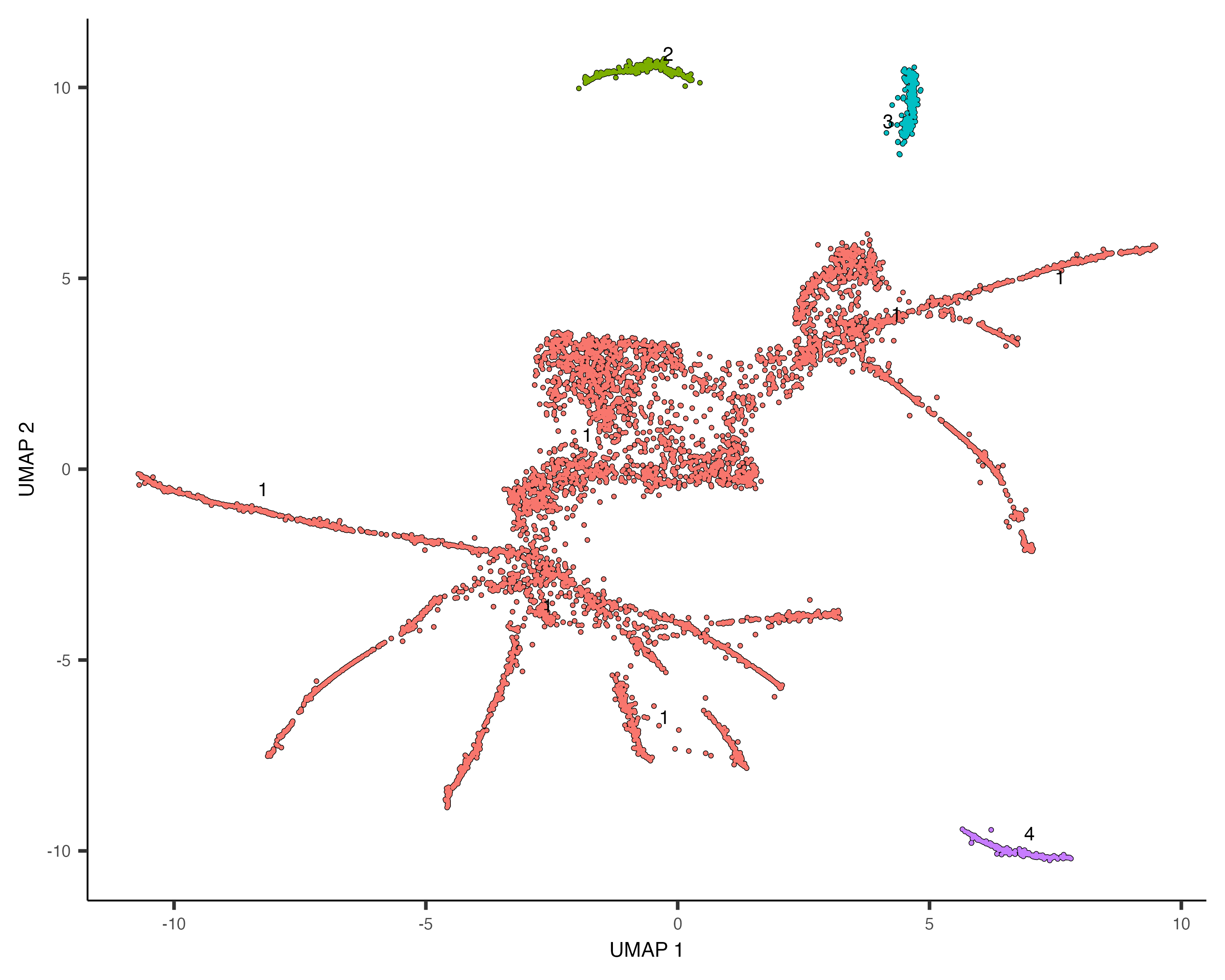
Learn the trajectory graph
Next, we will fit a learn_graph() function:
cds <- learn_graph(cds)
plot_cells(cds,
color_cells_by = "cell.type",
label_groups_by_cluster=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE)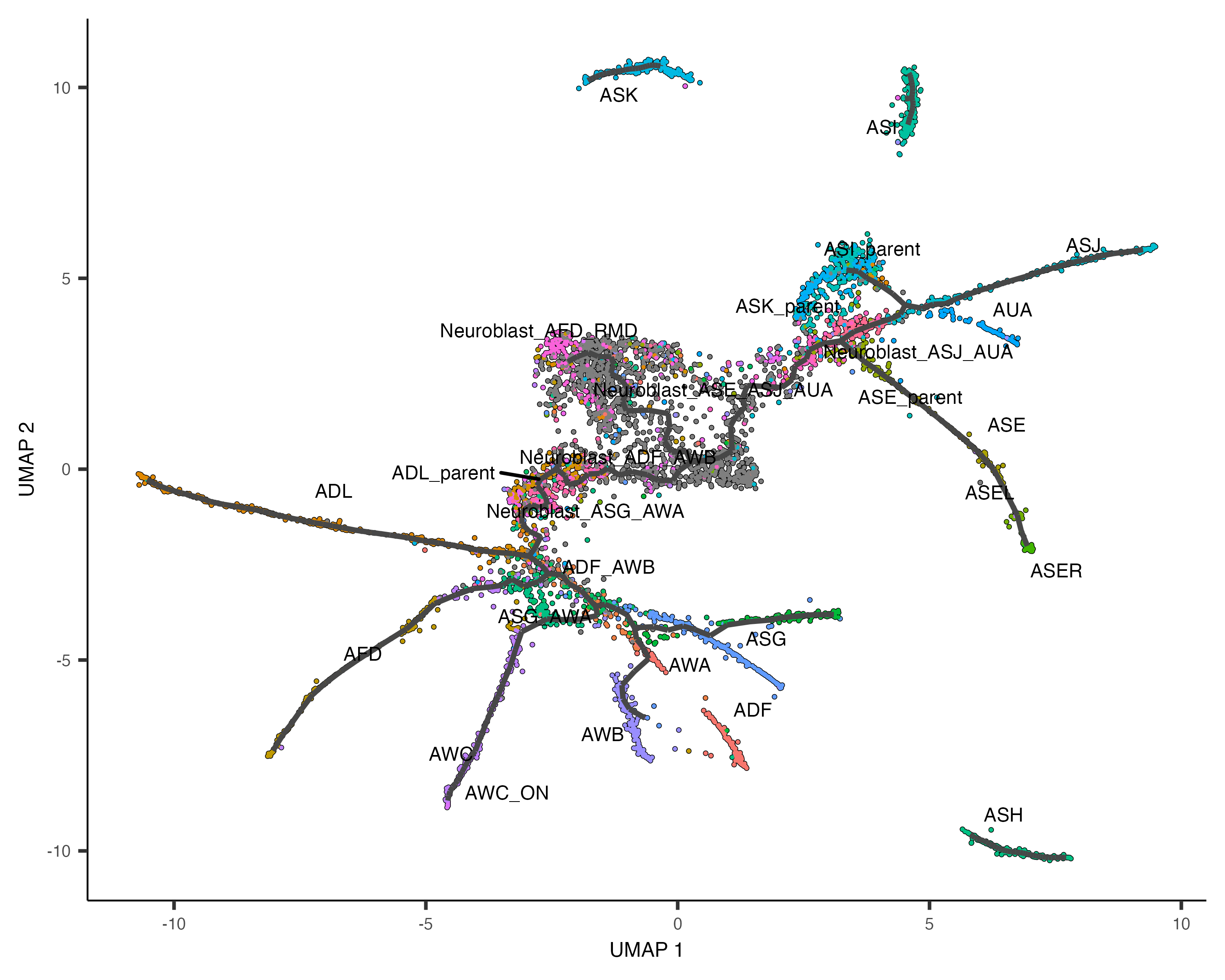
This graph will be used in many downstream steps, such as branch analysis and differential expression.
Order the cells in pseudotime
Once we've learned a graph, we are ready to order the cells according to their progress through the developmental
program. Monocle measures this progress in
What is pseudotime?
In many biological processes, cells do not progress in perfect synchrony. In single-cell expression studies of processes such as cell differentiation, captured cells might be widely distributed in terms of progress. That is, in a population of cells captured at exactly the same time, some cells might be far along, while others might not yet even have begun the process. This asynchrony creates major problems when you want to understand the sequence of regulatory changes that occur as cells transition from one state to the next. Tracking the expression across cells captured at the same time produces a very compressed sense of a gene's kinetics, and the apparent variability of that gene's expression will be very high.
By ordering each cell according to its progress along a learned trajectory, Monocle alleviates the problems that arise due to asynchrony. Instead of tracking changes in expression as a function of time, Monocle tracks changes as a function of progress along the trajectory, which we term "pseudotime". Pseudotime is an abstract unit of progress: it's simply the distance between a cell and the start of the trajectory, measured along the shortest path. The trajectory's total length is defined in terms of the total amount of transcriptional change that a cell undergoes as it moves from the starting state to the end state.
In order to place the cells in order, we need to tell Monocle where the "beginning" of the biological process is. We do so by choosing regions of the graph that we mark as "roots" of the trajectory. In time series experiments, this can usually be accomplished by finding spots in the UMAP space that are occupied by cells from early time points:
plot_cells(cds,
color_cells_by = "embryo.time.bin",
label_cell_groups=FALSE,
label_leaves=TRUE,
label_branch_points=TRUE,
graph_label_size=1.5)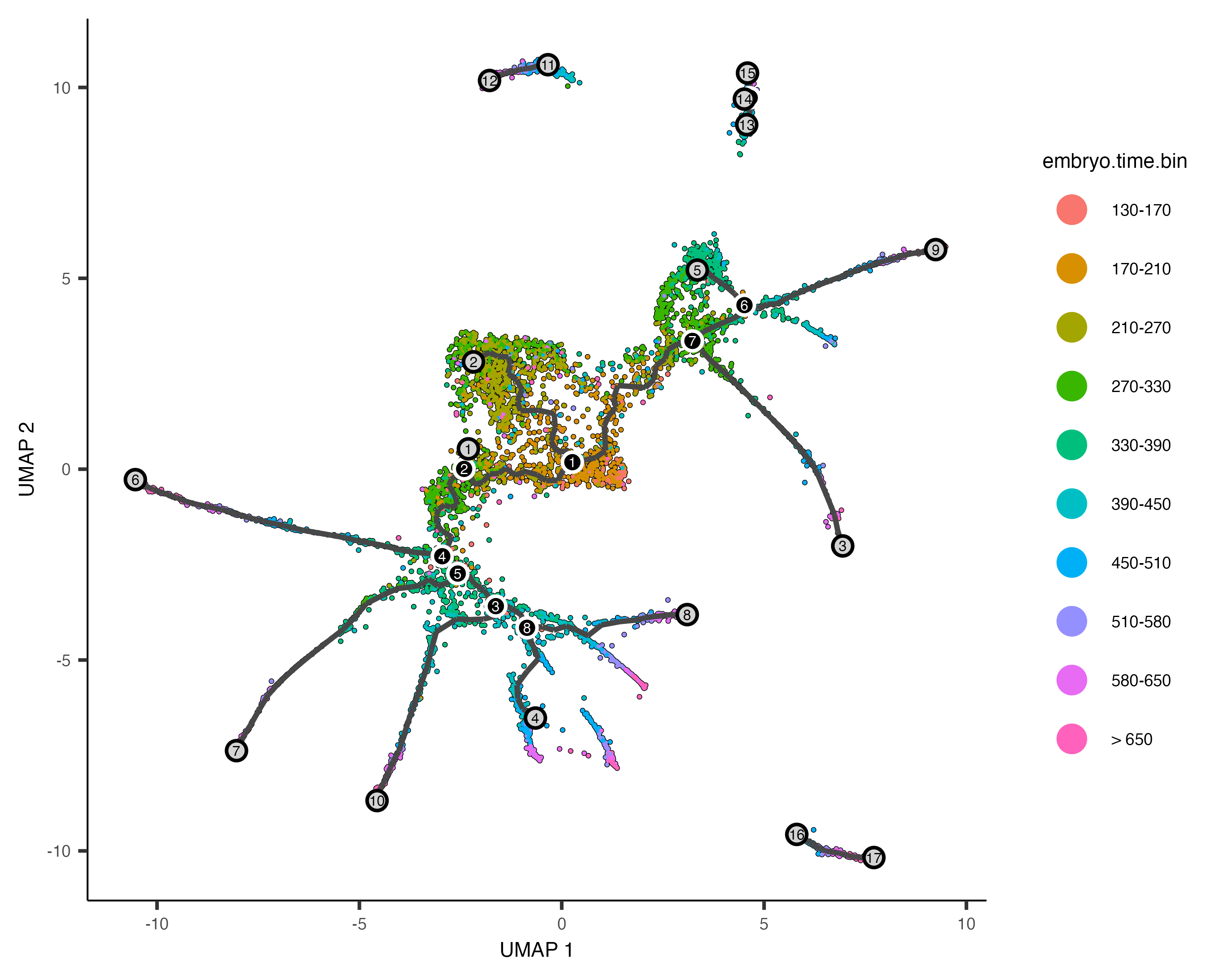
The black lines show the structure of the graph. Note that the graph is not fully connected: cells in different partitions
are in distinct components of the graph. The circles with numbers in them denote special points within the graph. Each
label_leaves and label_branch_points arguments to
plot_cells. Please note that numbers within the circles are provided for reference purposes only.
Now that we have a sense of where the early cells fall, we can call order_cells(), which will calculate where
each cell falls in pseudotime. In order to do so order_cells()needs you to specify the
cds <- order_cells(cds)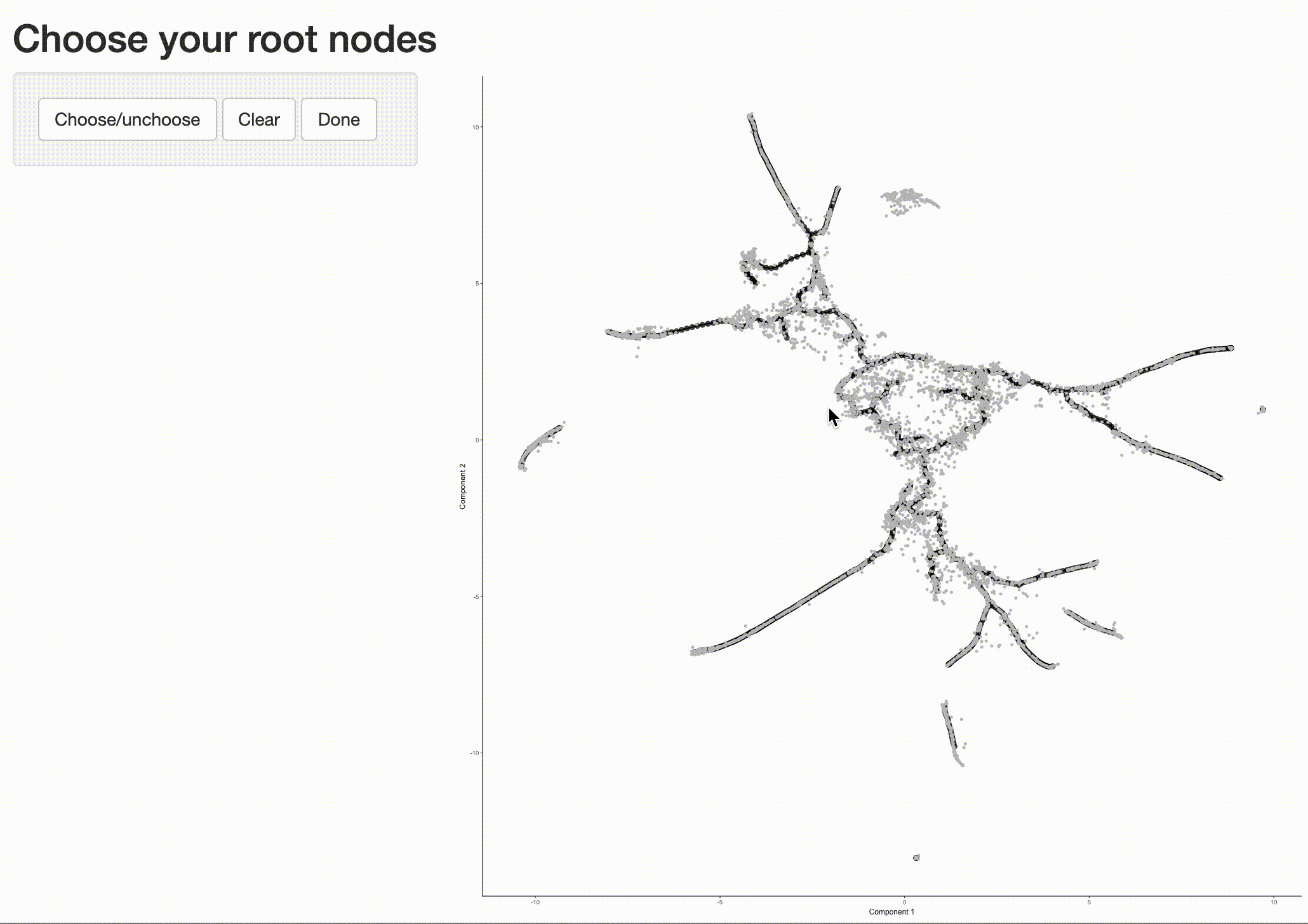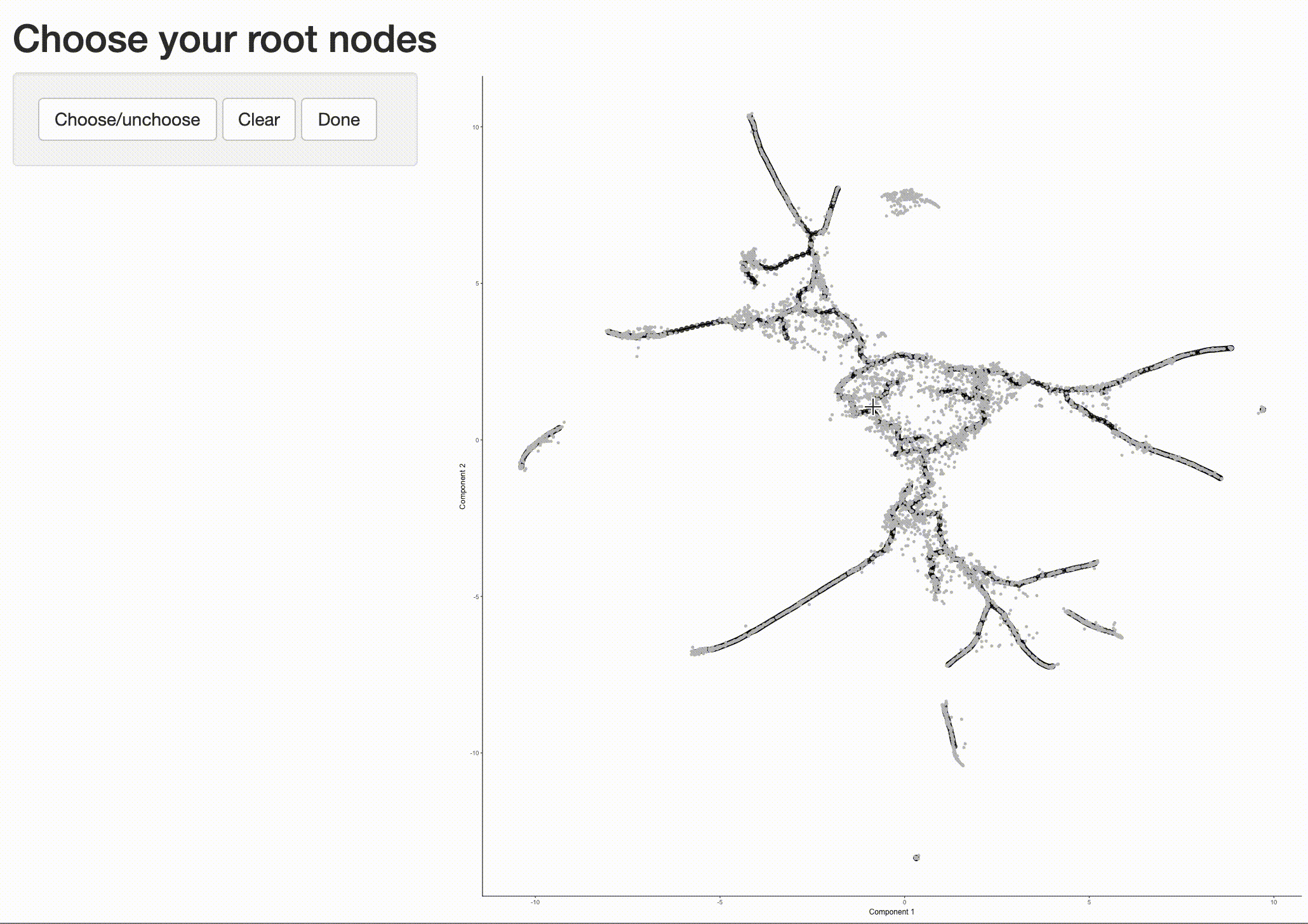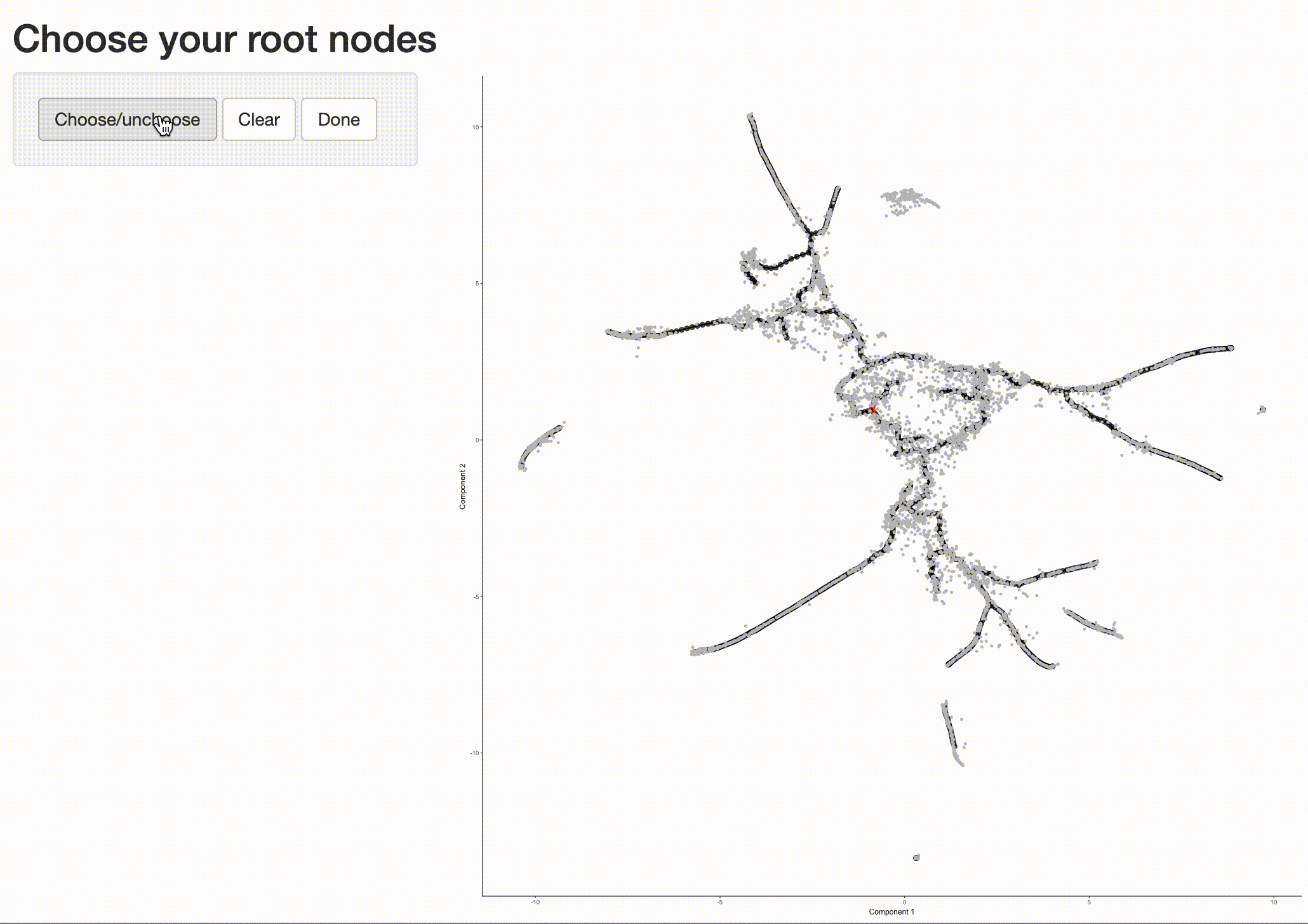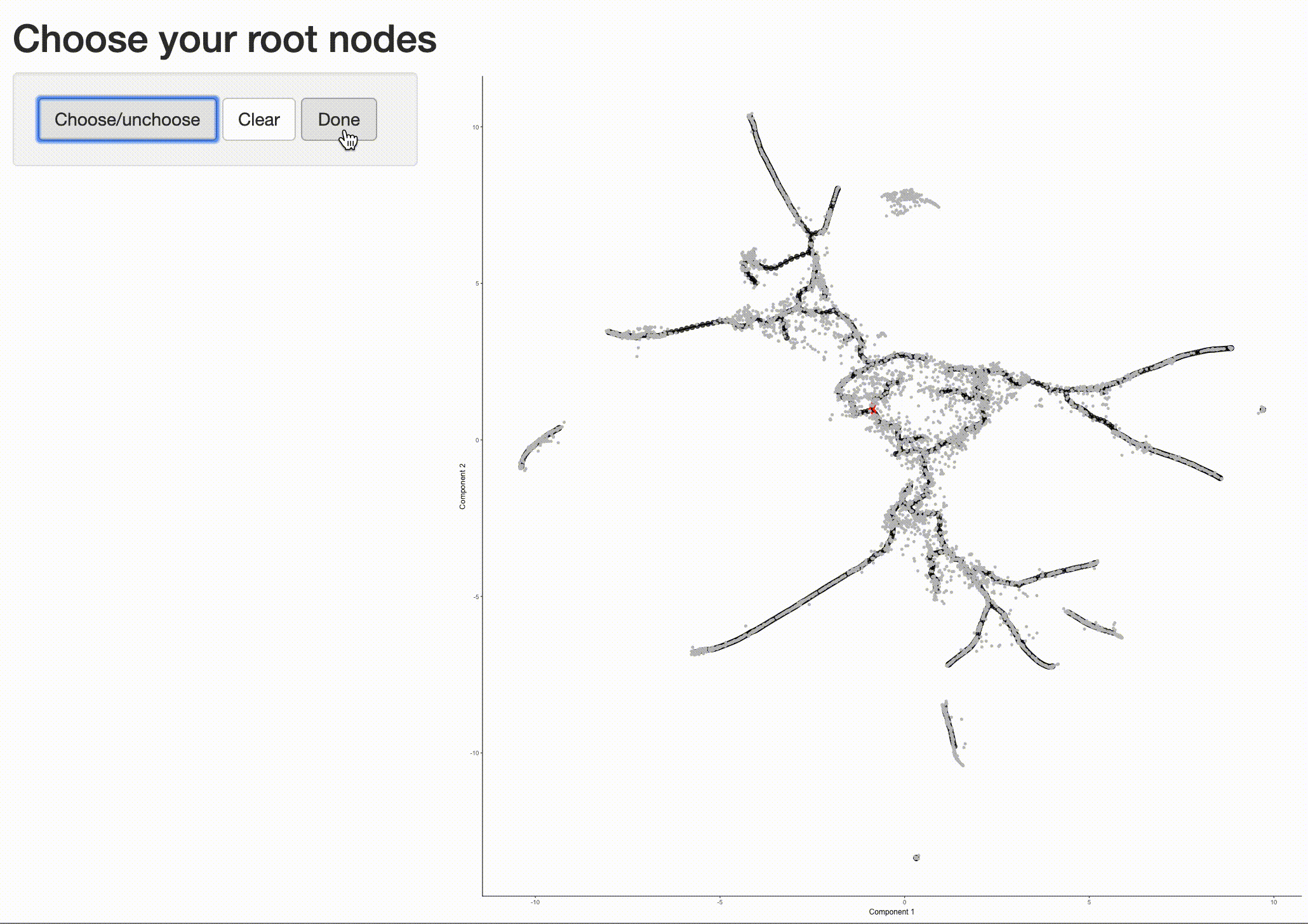
In the above example, we just chose one location, but you could pick as many as you want. Plotting the cells and coloring them by pseudotime shows how they were ordered:
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5)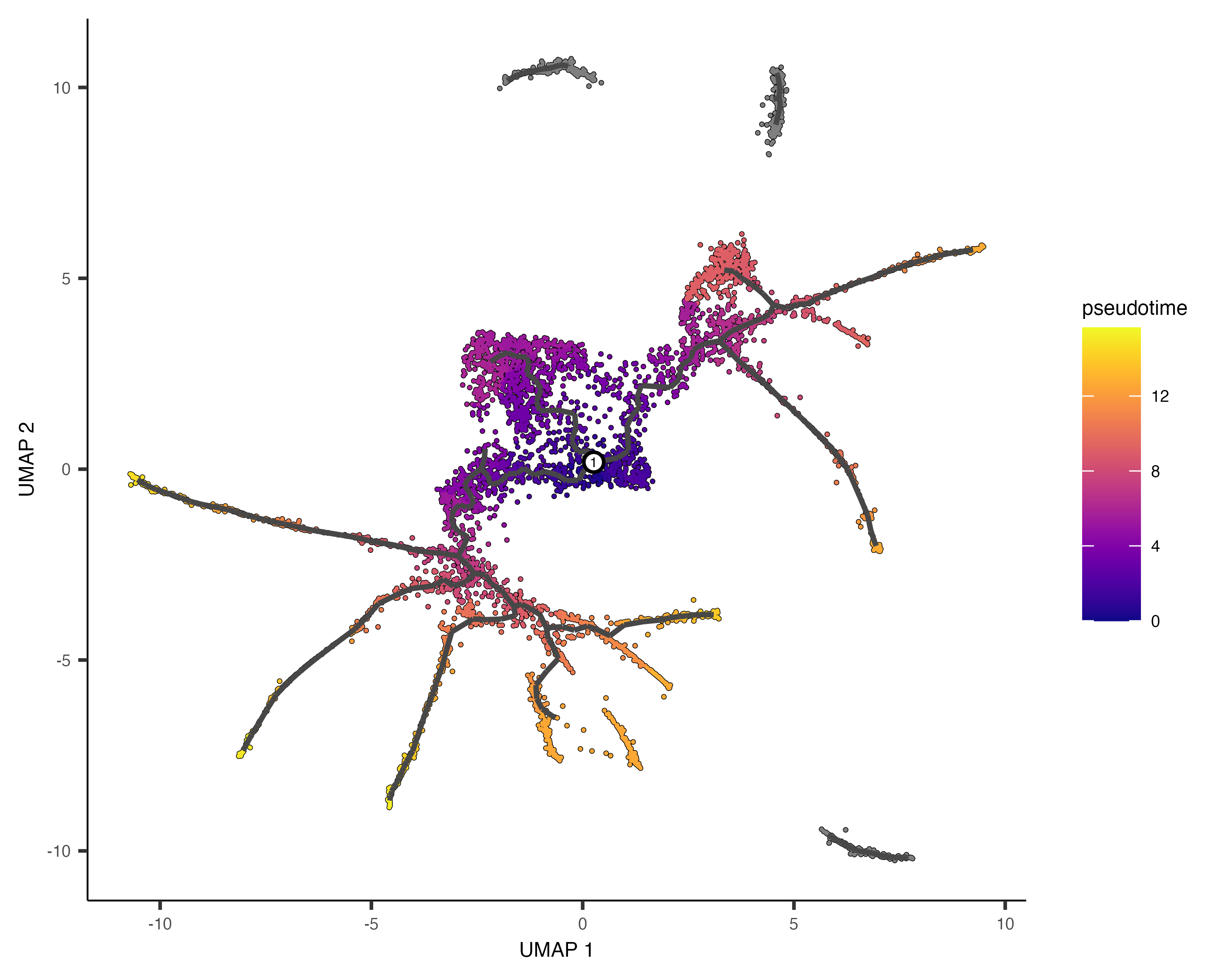
Note that some of the cells are gray. This means they have
It's often desirable to specify the root of the trajectory programmatically, rather than manually picking it. The function below does so by first grouping the cells according to which trajectory graph node they are nearest to. Then, it calculates what fraction of the cells at each node come from the earliest time point. Then it picks the node that is most heavily occupied by early cells and returns that as the root.
# a helper function to identify the root principal points:
get_earliest_principal_node <- function(cds, time_bin="130-170"){
cell_ids <- which(colData(cds)[, "embryo.time.bin"] == time_bin)
closest_vertex <-
cds@principal_graph_aux[["UMAP"]]$pr_graph_cell_proj_closest_vertex
closest_vertex <- as.matrix(closest_vertex[colnames(cds), ])
root_pr_nodes <-
igraph::V(principal_graph(cds)[["UMAP"]])$name[as.numeric(names
(which.max(table(closest_vertex[cell_ids,]))))]
root_pr_nodes
}
cds <- order_cells(cds, root_pr_nodes=get_earliest_principal_node(cds))
Passing the programatically selected root node to order_cells() via the root_pr_nodeargument
yields:
plot_cells(cds,
color_cells_by = "pseudotime",
label_cell_groups=FALSE,
label_leaves=FALSE,
label_branch_points=FALSE,
graph_label_size=1.5)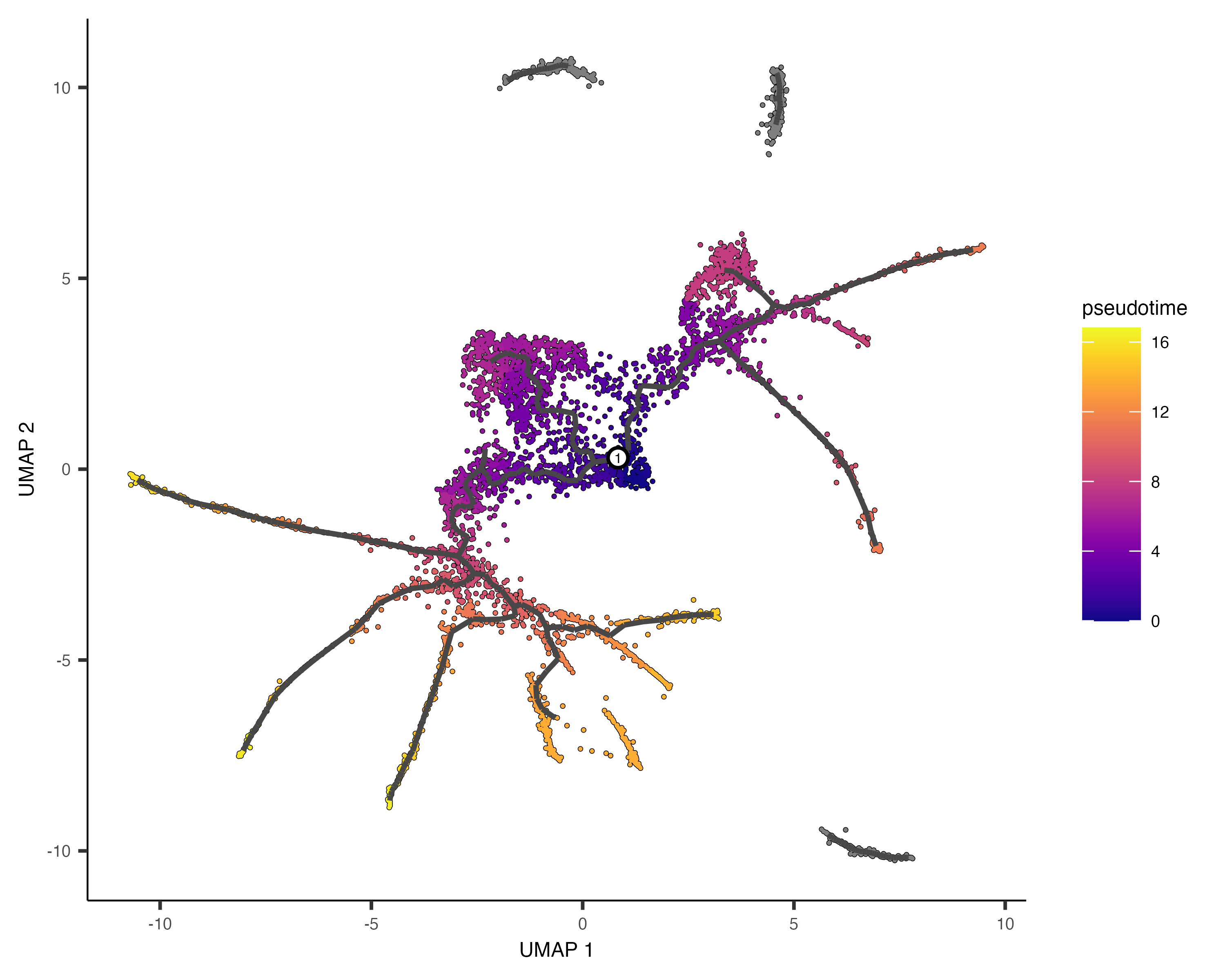
Note that we could easily do this on a per-partition basis by first grouping the cells by partition
using the partitions() function. This would result in all cells being assigned a finite pseudotime.
Subset cells by branch
It is often useful to subset cells based on their branch in the trajectory. The function choose_graph_segments
allows you to do so interactively.
cds_sub <- choose_graph_segments(cds)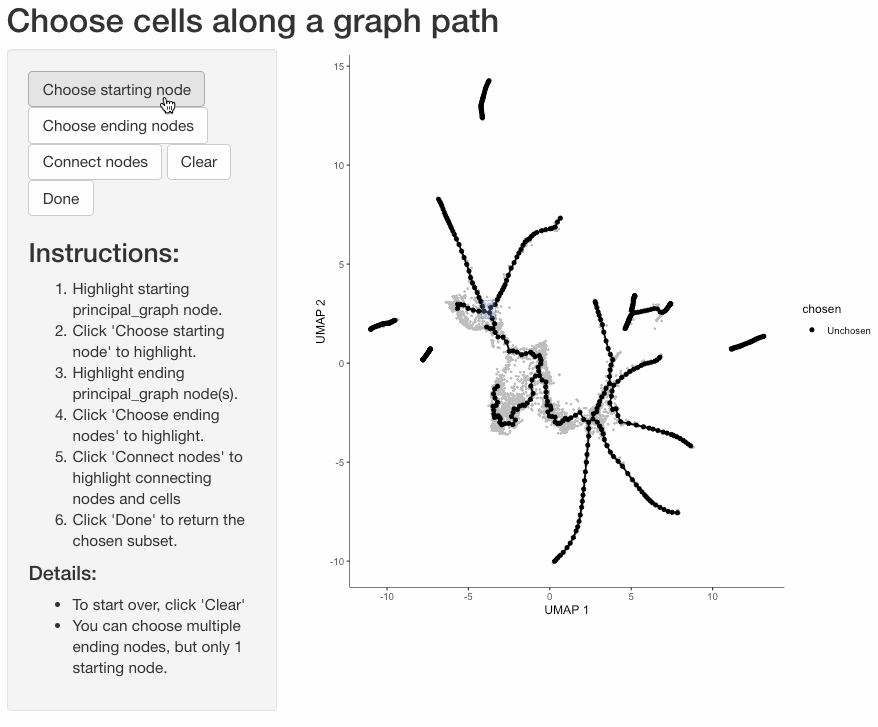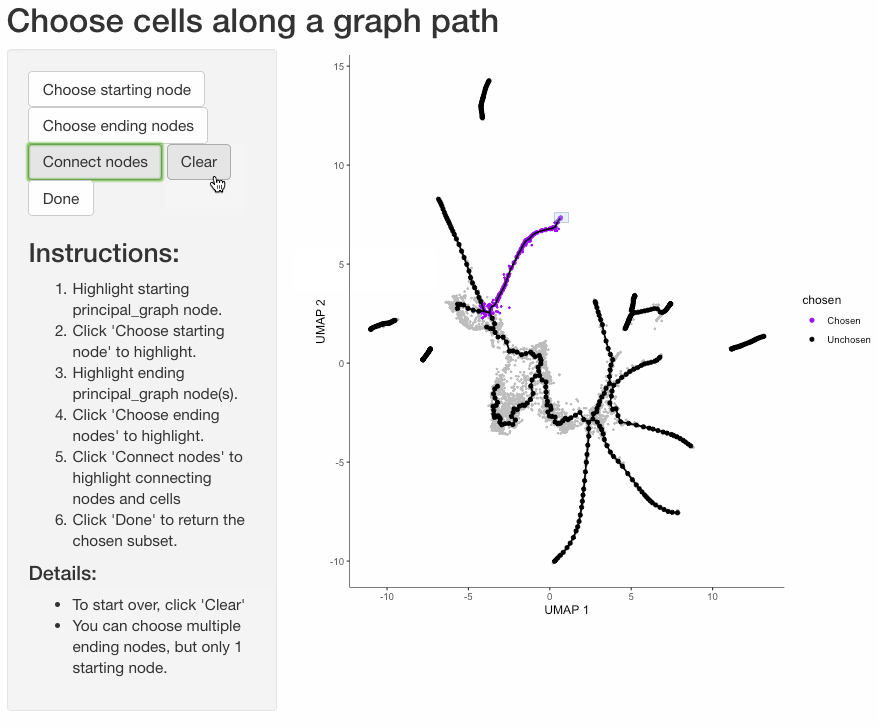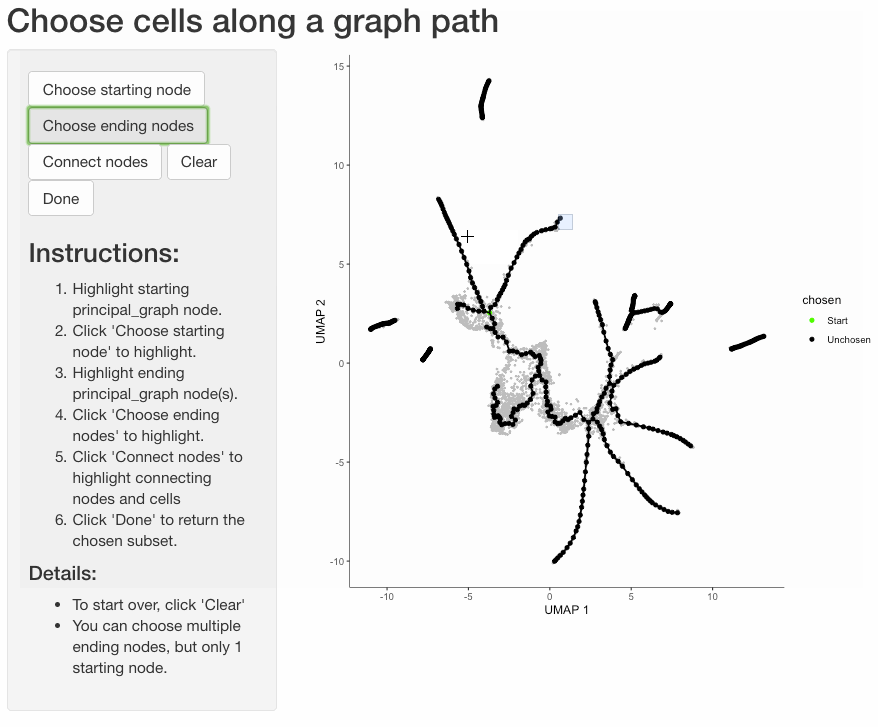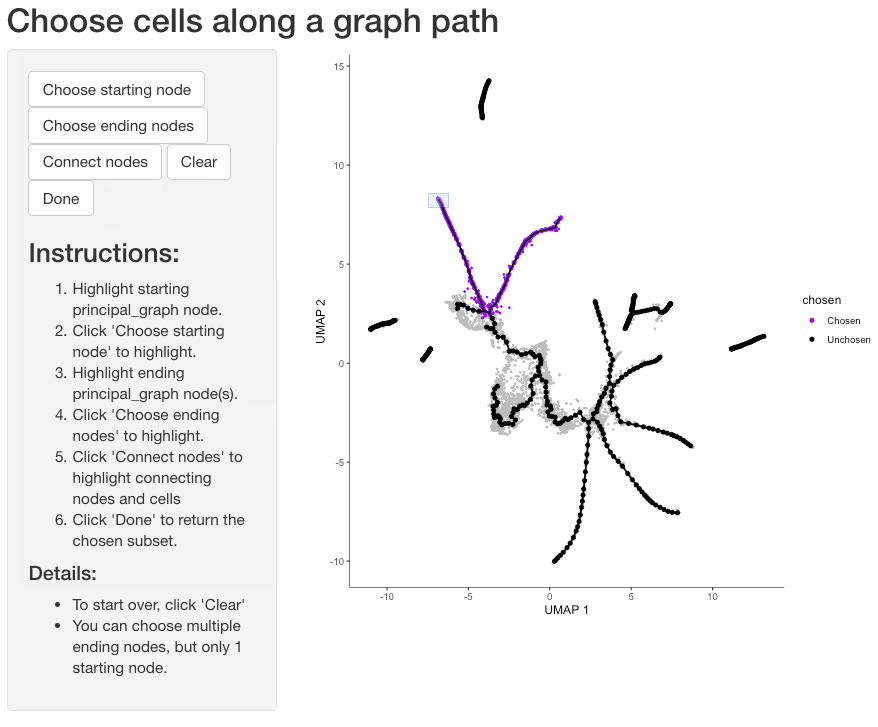
Working with 3D trajectories
cds_3d <- reduce_dimension(cds, max_components = 3)
cds_3d <- cluster_cells(cds_3d)
cds_3d <- learn_graph(cds_3d)
cds_3d <- order_cells(cds_3d, root_pr_nodes=get_earliest_principal_node(cds))
cds_3d_plot_obj <- plot_cells_3d(cds_3d, color_cells_by="partition")Previous Next