Monocle is an ongoing research project as well as a toolkit. If you use Monocle, please cite these papers in your work!
The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells
Cole Trapnell*, Davide Cacchiarelli*, Jonna Grimsby, Prapti Pokharel, Shuqiang Li, Michael Morse, Niall J. Lennon, Kenneth J. Livak, Tarjei S. Mikkelsen, John L. Rinn.
Nature Biotechnology 2014
Defining the transcriptional dynamics of a temporal process such as cell differentiation is challenging owing to the high variability in gene expression between individual cells. Time-series gene expression analyses of bulk cells have difficulty distinguishing early and late phases of a transcriptional cascade or identifying rare subpopulations of cells, and single-cell proteomic methods rely on a priori knowledge of key distinguishing markers. Here we describe Monocle, an unsupervised algorithm that increases the temporal resolution of transcriptome dynamics using single-cell RNA-Seq data collected at multiple time points. Applied to the differentiation of primary human myoblasts, Monocle revealed switch-like changes in expression of key regulatory factors, sequential waves of gene regulation, and expression of regulators that were not known to act in differentiation. We validated some of these predicted regulators in a loss-of function screen. Monocle can in principle be used to recover single-cell gene expression kinetics from a wide array of cellular processes, including differentiation, proliferation and oncogenic transformation
This is the original Monocle paper, which introduced the concept of pseudotime ordering for single-cell analysis
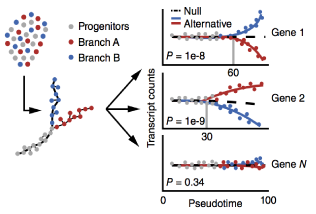
Single-cell mRNA quantification and differential analysis with Census
Xiaojie Qiu, Andrew Hill, Jonathan Packer, Dejun Lin, Yi-An Ma, Cole Trapnell
Nature Methods 2017
Single-cell gene expression studies promise to reveal rare cell types and cryptic states, but the high variability of single-cell RNA-seq measurements frustrates efforts to assay transcriptional differences between cells. We introduce the Census algorithm to convert relative RNA-seq expression levels into relative transcript counts without the need for experimental spike-in controls. Analyzing changes in relative transcript counts led to dramatic improvements in accuracy compared to normalized read counts and enabled new statistical tests for identifying developmentally regulated genes. Census counts can be analyzed with widely used regression techniques to reveal changes in cell-fate-dependent gene expression, splicing patterns and allelic imbalances. We reanalyzed single-cell data from several developmental and disease studies, and demonstrate that Census enabled robust analysis at multiple layers of gene regulation. Census is freely available through our updated single-cell analysis toolkit, Monocle 2.
This paper describes BEAM (used in branch analysis) and Census (the core of relative2abs)
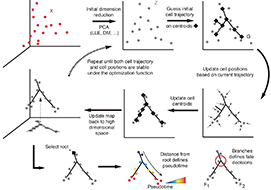
Reversed graph embedding resolves complex single-cell developmental trajectories
Xiaojie Qiu, Qi Mao, Ying Tang, Li Wang, Raghav Chawla, Hannah Pliner, Cole Trapnell
bioRxiv preprint
Organizing single cells along a developmental trajectory has emerged as a powerful tool for understanding how gene regulation governs cell fate decisions. However, learning the structure of complex single-cell trajectories with two or more branches remains a challenging computational problem. We present Monocle 2, which uses reversed graph embedding to reconstruct single-cell trajectories in a fully unsupervised manner. Monocle 2 learns an explicit principal graph to describe the data, greatly improving the robustness and accuracy of its trajectories compared to other algorithms. Monocle 2 uncovered a new, alternative cell fate in what we previously reported to be a linear trajectory for differentiating myoblasts. We also reconstruct branched trajectories for two studies of blood development, and show that loss of function mutations in key lineage transcription factors diverts cells to alternative branches on the a trajectory. Monocle 2 is thus a powerful tool for analyzing cell fate decisions with single-cell genomics.
This paper describes Monocle 2 and the use of Reversed Graph Embedding for single-cell analysis.